Oncoscience
As expected, based on rapamycin-like p53-mediated gerosuppression, mTOR inhibition acts as a checkpoint in p53-mediated tumor suppression
Mikhail V. Blagosklonny1
1 Roswell Park Comprehensive Cancer Center, Buffalo, NY 14263, USA
Correspondence to: Mikhail V. Blagosklonny, email: [email protected], [email protected]
Keywords: senescence; geroconversion; p53; sirolimus; cancer; hyperfunction theory
Received: August 20, 2022
Accepted: August 25, 2022
Published: August 30, 2022
Copyright: © 2022 Blagosklonny. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Recent work by Gu and co-workers (Kon et al., published in 2021), entitled “mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression”, seemingly “unexpectedly” demonstrated in mice that the ability of p53 to suppress mTOR is essential for tumor suppression early in life [1]. This actually was predicted in 2012 in the commentary entitled “Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression?” [2] [Note: rapalogs are rapamycin analogs]. The commentary [2] was written on another fascinating paper by the same senior author Gu and co-workers (Li et al.) “Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence” [3].
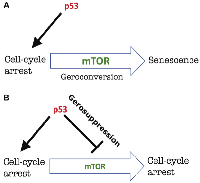
Mutant p53 (p53-3KR), constructed by Li et al., lacking all three then-known tumor-suppressing activities, still suppressed tumors [3]. To be precise, as noticed in the commentary [2], there were only two, not three, independent tumor-suppressing activities of p53 known at that time: namely, (i) apoptosis and (ii) cell-cycle arrest/senescence. Wild-type p53 does not directly induce the senescent phenotype; it induces cell-cycle arrest, which then converts to senescence (geroconversion) without any p53 assistance (Figure 1).
When the cell cycle gets arrested by any means (by p53, p21 or anything else), the arrested cell is not yet senescent at first. It will take several days (at least) in cell culture to observe senescent phenotype, including large cell morphology, Senescence-Associated Secretory Phenotype (SASP) and beta-Gal-staining (Figure 1). Geroconversion is driven by growth-promoting pathways such as mTOR and MAPK [4]. In fact, rapamycin and anything that inhibits mTOR such as serum-starvation, contact inhibition and anoxia partially suppresses geroconversion and the senescent phenotype (see for ref. [4]).
Then how does p53 causes senescence? It causes cell-cycle arrest, which, in growth-factor rich cell culture, may automatically lead to a senescent phenotype (Figure 2A).
[In analogy, a key to your home seemingly has two activities unlock the door and open the door. Yet, it only unlocks the door. When the door is unlocked by the key, you (or the wind) may open the door without key. But if an altered ”mutant” key cannot unlock the door, it cannot help to open it either].
Since mutant p53 (p53-3KR) cannot cause cell-cycle arrest, it cannot cause senescence either. On another hand, p53 may inhibit senescence by inhibiting mTOR-driven geroconversion [5, 6]. p53 inhibits mTOR [5–8]. When p53 causes quiescence (reversible arrest), it does so by inhibiting geroconversion (Figure 2B). The experimental confirmation is described [5] and discussed elsewhere [2, 9], so I will not discuss it here.
In agreement with in vitro results, it was shown that p53-null mice have increased mTOR activity [10], and that observation was confirmed by Kon et al., [1]. Rapamycin also delays cancer and increases lifespan in p53+/− mice [11, 12].
Kon et al., constructed a p53 mutant (p53-5KR) that is unable to inhibit mTOR [1]. Kon et al., showed that loss of mTOR inhibition led to inability to suppress tumors early in life. This defect was mitigated/reversed by treatment with rapamycin, further supporting the role of mTOR-inhibition in a cancer checkpoint [1]. As shown previously, rapamycin delayed tumorigenesis and extended lifespan in p53-null mice [13], an observation also confirmed by Kon et al., [1].
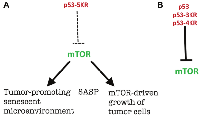
As anticipated in 2012, in the absence of rapamycin-like p53-mediated gerosuppression, mTOR favors a senescent microenvironment [2], associated with cancer promotion [14–17]. SASP is one of the mechanisms of tumor-promotion by senescent cells and is inhibited by both rapamycin and p53 [12, 18–20]. Rapamycin decreases the ability of microenvironment and tumor stroma to promote cancer growth [12, 15, 16]. Also, mTOR may directly increase pre-cancer/cancer cell growth (Figure 3).
To keep the focus on the mTOR story, which was unexpected for Kan et al., [1], I did not discuss the fourth anti-cancer activity of p53 discovered by Gu and co-workers: ferroptosis [1, 21, 22]. I still must mention this fascinating story because p53-5KD should be compared with p53-4KD, not with p53-3KD. Mutant p53-4KR, which lacks the ability to undergo p53-mediated cell cycle arrest/senescence, apoptosis, and ferroptosis, retains the ability to inhibit mTOR activity, while this activity is completely abolished in p53-5KR [1]. This work by Gu and co-workers adds a fourth activity (ferroptosis) [1, 21, 22] to three anti-cancer activities proposed in 2012 [2]: cycle arrest, apoptosis, and rapamycin-like gerosuppression or, in simple words, mTOR inhibition [2].
CONFLICTS OF INTEREST
Author has no conflicts of interest to declare.
- 1. mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Genes Dev. 2021; 35: 59-64. https://doi.org/10.1101/gad.340919.120. PMID:33303641
- 2. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY). 2012; 4: 450-455. https://doi.org/10.18632/aging.100475. PMID:22869016
- 3. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012; 149: 1269-1283. https://doi.org/10.1016/j.cell.2012.04.026. PMID:22682249
- 4. Cell senescence, rapamycin and hyperfunction theory of aging. Cell Cycle. 2022; 21: 1456-1467. https://doi.org/10.1080/15384101.2022.2054636. PMID:35358003
- 5. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660-9664. https://doi.org/10.1073/pnas.1002298107. PMID:20457898
- 6. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2: 344-352. https://doi.org/10.18632/aging.100160. PMID:20606252
- 7. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005; 102: 8204-8209. https://doi.org/10.1073/pnas.0502857102. PMID:15928081
- 8. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134: 451-460. https://doi.org/10.1016/j.cell.2008.06.028. PMID:18692468
- 9. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010; 9: 4256-4257. https://doi.org/10.4161/cc.9.21.13785. PMID:20980826
- 10. Dysregulation of the mTOR pathway in p53-deficient mice. Cancer Biol Ther. 2013; 14: 1182-1188. https://doi.org/10.4161/cbt.26947. PMID:24184801
- 11. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY). 2012; 4: 709-714. https://doi.org/10.18632/aging.100498. PMID:23123616
- 12. p53 and rapamycin are additive. Oncotarget. 2015; 6: 15802-15813. https://doi.org/10.18632/oncotarget.4602. PMID:26158292
- 13. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4: 715-722. https://doi.org/10.18632/aging.100496. PMID:23117593
- 14. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5: 99-118. https://doi.org/10.1146/annurev-pathol-121808-102144. PMID:20078217
- 15. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012; 181: 278-293. https://doi.org/10.1016/j.ajpath.2012.03.017. PMID:22698676
- 16. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015; 17: 1049-1061. https://doi.org/10.1038/ncb3195. PMID:26147250
- 17. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013; 123: 966-972. https://doi.org/10.1172/JCI64098. PMID:23454759
- 18. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017; 16: 564-574. https://doi.org/10.1111/acel.12587. PMID:28371119
- 19. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep. 2018; 8: 2410. https://doi.org/10.1038/s41598-018-20000-4. PMID:29402901
- 20. Do p53 stress responses impact organismal aging? Transl Cancer Res. 2016; 5: 685-691. https://doi.org/10.21037/tcr.2016.12.02. PMID:30984573
- 21. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015; 520: 57-62. https://doi.org/10.1038/nature14344. PMID:25799988
- 22. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016; 17: 366-373. https://doi.org/10.1016/j.celrep.2016.09.022. PMID:27705786
 All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.